Project files provided as supporting information to the manuscript "Ligand-protein interactions in lysozyme investigated through a dual-resolution model"
Authors/Creators

- 1. Max Planck Institute for Polymer Research
- 2. University of Trento
Description
README file for the project files provided as supporting information to the manuscript "Ligand-protein interactions in lysozyme investigated through a dual-resolution model"
February 12, 2020
Authors: Raffaele Fiorentini, Kurt Kremer and Raffaello Potestio
================================
Overview
The dataset is organised in three (compressed) subfolders (see the tree diagrams in each section):
- annihilation
- decoupling
- density
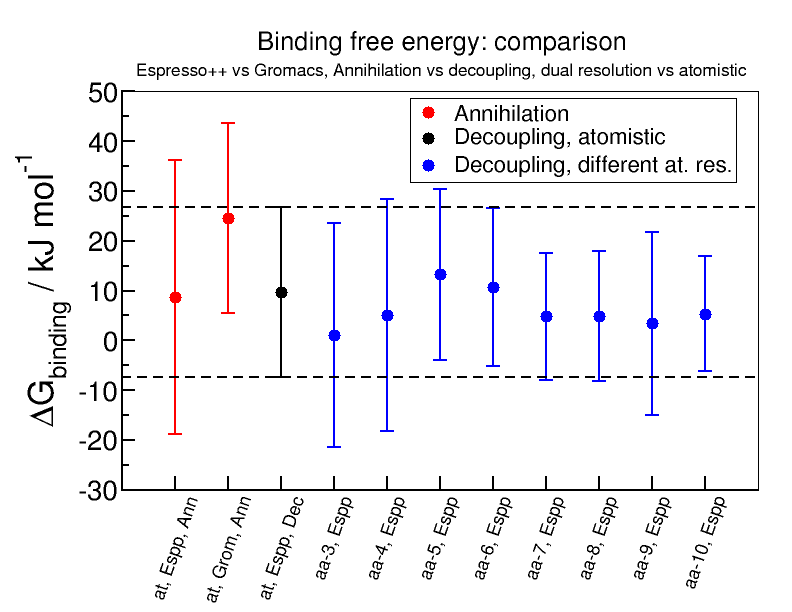
The figure deltaG_binding_ann_dec_comparison.png shows the results of binding free energy calculations comparing the values obtained both for annihilation and decoupling.
The figure deltaG_binding_annih_gromacs_espp.png displays the results for Binding FE, comparing the values obtained in GROMACS and ESPResSo++.
The README.pdf file contains detailed information about these folders and their content.
================================
The "annihilation" folder contains all results concerning the calculation of binding free energy in case of annihilation and it is divided in two parts:
- complex
- ligand
In "complex" are reported the results of Ligand-Protein FE both in ESPResSo++ and GROMACS. All simulations are fully-atomistic.
In "ligand" are reported the results of ligand solvation free energy both in ESPResSo++ and GROMACS. All simulations are fully-atomistic.
====
The "decoupling" folder contains all results concerning the calculation of binding free energy in case of decoupling and it is divided in three parts:
- complex-DualRes
- complex-FullyAT
- ligand
In "complex-DualRes" are reported the results of Ligand-Protein FE only in ESPResSo++ (GROMACS cannot do decoupling). The system is simulated in Dual-Resolution. It is possible to find the trajectory files in the sub-directories "lambdaindex-0" and "lambdaindex-30".
In "complex-fullyAT" are reported the results of Ligand-Protein FE only in ESPResSo++. The system simulated is fully-atomistic. It is possible to find the trajectory file in the sub-directories "lambdaindex-0" and "lambdaindex-30".
In "ligand" are reported the results of ligand solvation free energy only in ESPResSo++. All simulations are fully-atomistic. It is possible to find the trajectory file in the sub-directories "lambdaindex-0" and "lambdaindex-20".
====
The "density" folder contains the data for the tuning of the c parameter of the steric repulsion among residues. This parameter is tuned so that the water density attains the value computed in all-atom simulations.
Files
annihilation.zip
Files
(755.1 MB)
| Name | Size | Download all |
|---|---|---|
|
md5:60eae9c2591c46f061f02d21fd50ac2f
|
329.1 MB | Preview Download |
|
md5:c68c2ac891cff8b2f8b39aa447f75102
|
416.4 MB | Preview Download |
|
md5:e6578032c040312568439e353f706b8a
|
14.5 kB | Download |
|
md5:2900c2e58ee3781d6334a7243785dc78
|
15.7 kB | Preview Download |
|
md5:9e7226d8d1c633d4b7a37df895409b14
|
9.4 MB | Preview Download |
|
md5:b838eb72ff7e5ccd7f595b9979513184
|
131.5 kB | Preview Download |
{kind=link}
Additional details
Funding
References
- Fiorentini et al., Ligand-protein interactions in lysozyme investigated through a dual-resolution model, arXiv:2002.05263