Amyloid Illustrator
Authors/Creators

- 1. University of California Los Angeles
- 2. Molecular Biology Institute
Description
This collection of software, collectively called "Amyloid Illustrator" automatically creates illustrations of amyloid fibrils from a set of atomic coordinates (pdb format) supplied by the user. Examples of these illustrations can be viewed on our Amyloid Atlas web page. Amyloid Illustrator was originally installed in a server in August 2022 (https://srv.mbi.ucla.edu/AmyloidAtlas/Illustrator/); however, that service was interrupted in June 2024 due to hackers. Until the issue is resolved, I have made available the software here.
--------------INSTRUCTIONS-FOR-INSTALLATION----------------------
Here are instructions for installing Amyloid Illustrator on your Linux computer.
Please download the following 5 fortran code files, listed in order of use:
1) pdbclean.f removes hydrogen atoms, ANISOU records, and multiple conformations.
2) isolatefibrilasu_v04.f identifies the most buried layer in an amyloid fibril (.pdb) which is then used for illustration.
3) accessiblesurfacearea_v07.2u.f estimates the standard free energy from a .pdb file and atomic solvation parameters.
4) rotxz.f rotates the amyloid coordinates to attain the desired orientation for illustration.
5) autoschematic171.f creates illustrations of one amyloid layer: one colored by polarity, another colored by energy.
These files should be compiled and the executables placed together in a single directory which I refer to below as $exedir.
For example, on my local linux computer, I compile the 5 fortran codes using the following commands:
gfortran pdbclean_v02.f -o pdbclean_v02
gfortran isolatefibrilasu_v04.f -o isolatefibrilasu_v04
gfortran accessiblesurfacearea_v07.2u.f -o accessiblesurfacearea_v07.2u
gfortran rotxz.f -o rotxz
gfortran autoschematic171.f -o autoschematic171
Additionally, please download the script "autoillustrate.com" which calls the programs in the necessary order.
Dependencies: The end of the "autoillustrate.com" script calls upon the programs "convert" and "display" from ImageMagick (https://imagemagick.org/index.php). These programs are convenient for producing illustrations, but not essential. The program "convert" simply converts the postscript files output by Amyloid Illustrator to a more friendly png format, and "display" displays the illustration on the screen. There are other programs you can use to view postscript format files (e.g. Adobe Illustrator, Adobe Photoshop, ghostscript, gsview, etc.).
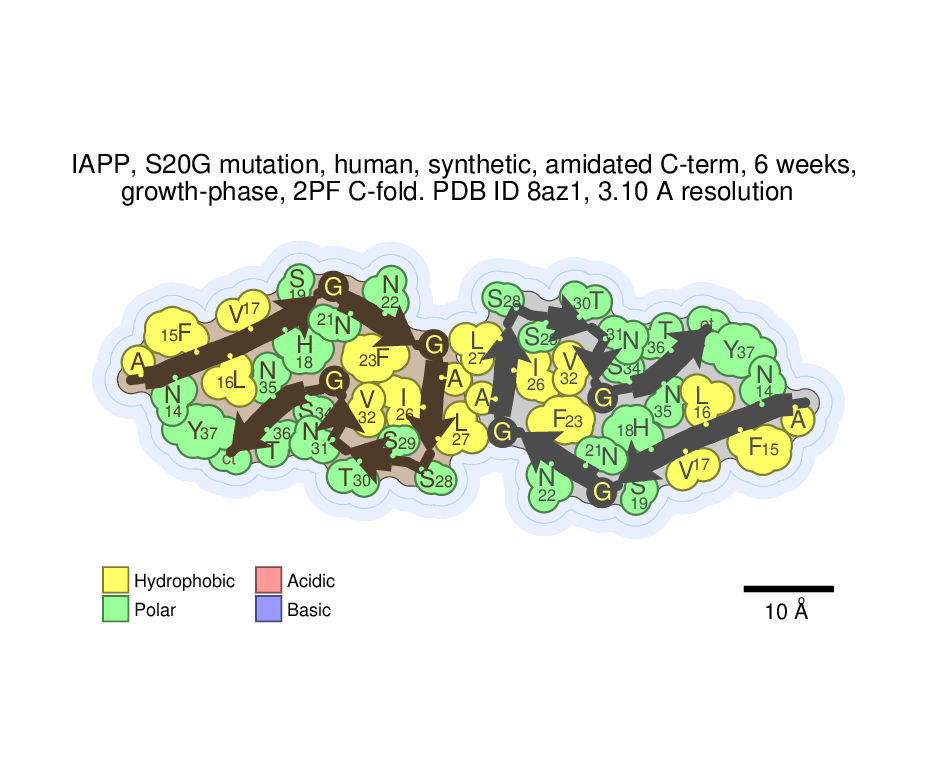
I suggest testing the installation on PDB ID code 8az1, an amyloid fibril of IAPP. The coordinates in pdb format can be downloaded from https://files.rcsb.org/download/8AZ1.pdb
Testing the installation requires some minor edits at the top of the script "autoillustrate.com".
The variable "exedir" specifies the directory where you put the executables that you compiled. If this isn't correct, the programs won't run.
The variable "pdbin" specifies the name of input coordinate file in pdb format. In this test case, the pdb file is "8AZ1.pdb"
The variable "title1" specifies a title to be printed at the top of the output illustrations (optional).
The variable "reso" specifies the resolution of the structure (optional).
The variable "pHvalu' specifies the pH value to use for the calculation (defines the charge state of ionizable side chains).
The variable "rotx" specifies the number of degrees to rotate the structure around the "x" axis. (usually 0 or 180)
The variable "rotz" specifies the number of degrees to rotate the structure around the "z" axis. (usually between -360 and +360).
No further edits below "rotz" are required
I suggest that you put the files "autoillustrate.com" and "8AZ1.pdb" in a new directory (not in $exedir). Running the script will produce about 31 output files, and that can become messy if produced in a directory already containing many files.
To produce the illustrations, just type "./autoillustrate.com". The output illustrations should be produced in files called "8AZ1_polarimap.ps" and "8AZ1_energymap.ps", in this case.
The output illustrations should look like this: https://people.mbi.ucla.edu/sawaya/amyloidatlas/residuefull/8az1_polarimap.png and this https://people.mbi.ucla.edu/sawaya/amyloidatlas/energyfull/8az1_energymap.png .
{kind=link}
{kind=link}
-----------------NOTES-ON-USING-AMYLOID-ILLUSTRATOR-------------------
1) Supply coordintes in .pdb format; the program does not handle mmcif format.
2) Atoms missing from any residue will cause the job to crash. Build in any missing atoms or delete the residue completely (e.g. use COOT).
3) If you are submitting a structure determined by NMR, submit only a single model of interest. Multiple MODELs (e.g. PDB ID 2beg) will produce errors.
4) Include coordinates for at least three layers of the amyloid fibril. I regard a "layer" as a slice of the fibril perpendicular to the fibril axis. More specifically, one layer includes all atoms in the asymmetric unit of the fibril assuming C1 helical symmetry. It is important to include multiple layers to get an accurate estimate of the energetic stability of the fibril. Our method of evaluating energetic stability depends on measuring surface area buried by the fibril assembly. If you supply only one layer, the energetic stability of the fibril will be greatly underestimated. Note that most amyloid fibril models deposited in the PDB have at least 3 layers, and so work fine with this server. The server will report the energy for the layer most buried in the fibril.
5) The coordinate file should contain no more than 9000 atoms, otherwise the job will crash.
6) No two atoms in the .pdb file should share the same chain ID and residue number (this is normally the case).
"title1" can be set to any descriptive text you like. The title will be displayed and centered at the top of the schematic. If you don't want a title, then enter blank spaces for "title1". "title1" must not exceed 100 characters.
"rotx" corresponds to rotation (degrees) of the amyloid fibril around the "x" axis (horizontal axis).
"rotx" is typically set to either 0 or 180 degrees, corresponding to views of the fibril from either the +z or -z directions.
Any value from -180 to +180 is allowable, but in most cases, 0 or 180 are the most informative. It is standard for the fibril axis to be oriented parallel to the "z" axis in coordinate files determined by CryoEM, but not for NMR structures. The orientation of the fibril axis does not effect the energetic evaluation, but it will change the perspective of the fibril rendered in the output figure. If the perspective in the figure looks strange (i.e. small and overly crowded with atoms), check whether the fibril axis is parallel to "z". If not, use a program such as COOT to reorient the fibril axis to be parallel with "z" and submit the reoriented coordinates.
"rotz" corresponds to rotation (degrees) of the amyloid fibril around the "z" axis (perpendicular to the screen).
"rotz" can have any value (-360 to +360). It adjusts the orientation of the fibril in the plane of the screen.
"pHvalu" is the value of pH used for the energy calculation.
The pH value given here will affect the charge assigned to ionizable residues. The ionization state will affect the atomic solvation parameter assigned to an atom, and thus its energy.
----------------------------TROUBLESHOOTING----------------------------------
If there are missing atoms, the script will crash.
The error can be identified by grepping the file accessiblesurfacearea_v07.2i.log
and it will have a message like this:
Missing atoms from A 15 PHE 10
The server should report that PHE 15 from chain A is missing atoms.
If there is an error due to too many atoms, the script will crash.
The error can be identified by grepping the file accessiblesurfacearea_v07.2i.log
and it will have a message like this:
Redimension for more than 9000 atoms.
Michael R. Sawaya
Notes
Files
Files
(318.3 kB)
| Name | Size | Download all |
|---|---|---|
|
md5:de20104cae1a9fb6cd43a2b2077971c4
|
112.6 kB | Download |
|
md5:e06965a2aa6c135f73b3b0d9052b14c4
|
4.7 kB | Download |
|
md5:c7250020f1d41858424514ac84ca3664
|
171.8 kB | Download |
|
md5:63f8d5f682c91879b01a85bce5643367
|
17.3 kB | Download |
|
md5:0a404bd84605d156c649b9d3a5b0494d
|
8.1 kB | Download |
|
md5:7d70d04204c33c62d7a5601ba6e5b8cb
|
3.8 kB | Download |
Additional details
Related works
- Compiles
- Dataset: https://people.mbi.ucla.edu/sawaya/amyloidatlas/ (URL)
- Describes
- Journal article: 10.1016/j.cell.2021.08.013 (DOI)
Funding
Dates
- Submitted
-
2025-04-14