vAMPirus: An automated, comprehensive virus amplicon sequencing analysis program
Authors/Creators

- 1. Rice University
- 2. Ludwig-Maximilians-University of Munich
Description
Abstract
Here we present vAMPirus (https://github.com/Aveglia/vAMPirus.git), an automated and easy-to-use virus amplicon sequencing analysis program. Recent advances in sequencing approaches and technology have revealed the astounding diversity of viruses in natural environments. Amplicon sequencing is an effective approach for identifying genetic variants within a specific viral group or population. Although, the high volume of amplicon data produced combined with the high mutation rates across different viral genes can make it difficult to scale and standardize analytical approaches. vAMPirus is an accessible automated virus amplicon sequencing analysis program integrated within the Nextflow framework, which allows users to tailor analyses to their data, and can then be easily scaled and standardized across datasets. The vAMPirus program contains two different analytical modes: a “DataCheck” mode and an “Analyze” mode. In the DataCheck mode, vAMPirus generates an interactive html report file containing information regarding sequencing success per sample, as well as a preliminary look at the clustering behavior of the data, which can be leveraged to inform future analyses. The Analyze mode conducts a comprehensive analysis of the data, generating a wide range of results and outputs, including an interactive report with figures and statistics. We anticipate that vAMPirus will benefit the virology community by promoting accessibility to in-depth analyses and the standardization of analytical practices, facilitating reproducibility and cross-study comparisons.
Brief description
Viruses are the most abundant biological entities on the planet and with advances in next-generation sequencing technologies, there has been significant effort in deciphering the global virome and its impact in nature (Suttle 2007; Breitbart 2019). A common method for studying viruses in the lab or environment is amplicon sequencing, an economic and effective approach for investigating virus diversity and community dynamics. The highly targeted nature of amplicon sequencing allows in-depth characterization of genetic variants within specific viral groups facilitating both virus discovery and screening within samples. Although, the high volume of amplicon data produced combined with the highly variable nature of virus evolution across different genes and virus-types can make it difficult to scale and standardize analytical approaches. To address this, we present vAMPirus (https://github.com/Aveglia/vAMPirus.git), an automated and comprehensive virus amplicon sequencing analysis program that is integrated with the Nextflow scientific workflow manager facilitating easy scalability and standardization of analyses (Nextflow.io). vAMPirus was also designed to be accessible, being composed completely of open-source software and having thorough help documentation with step-by-step instructions to install and run vAMPirus on diverse operating systems.
This is a very brief write-up of vAMPirus and its capabilities, please see the manual for a more detailed description of each process within the mentioned analytical modes - https://bit.ly/2M1nDdw
The two vAMPirus analytical modes
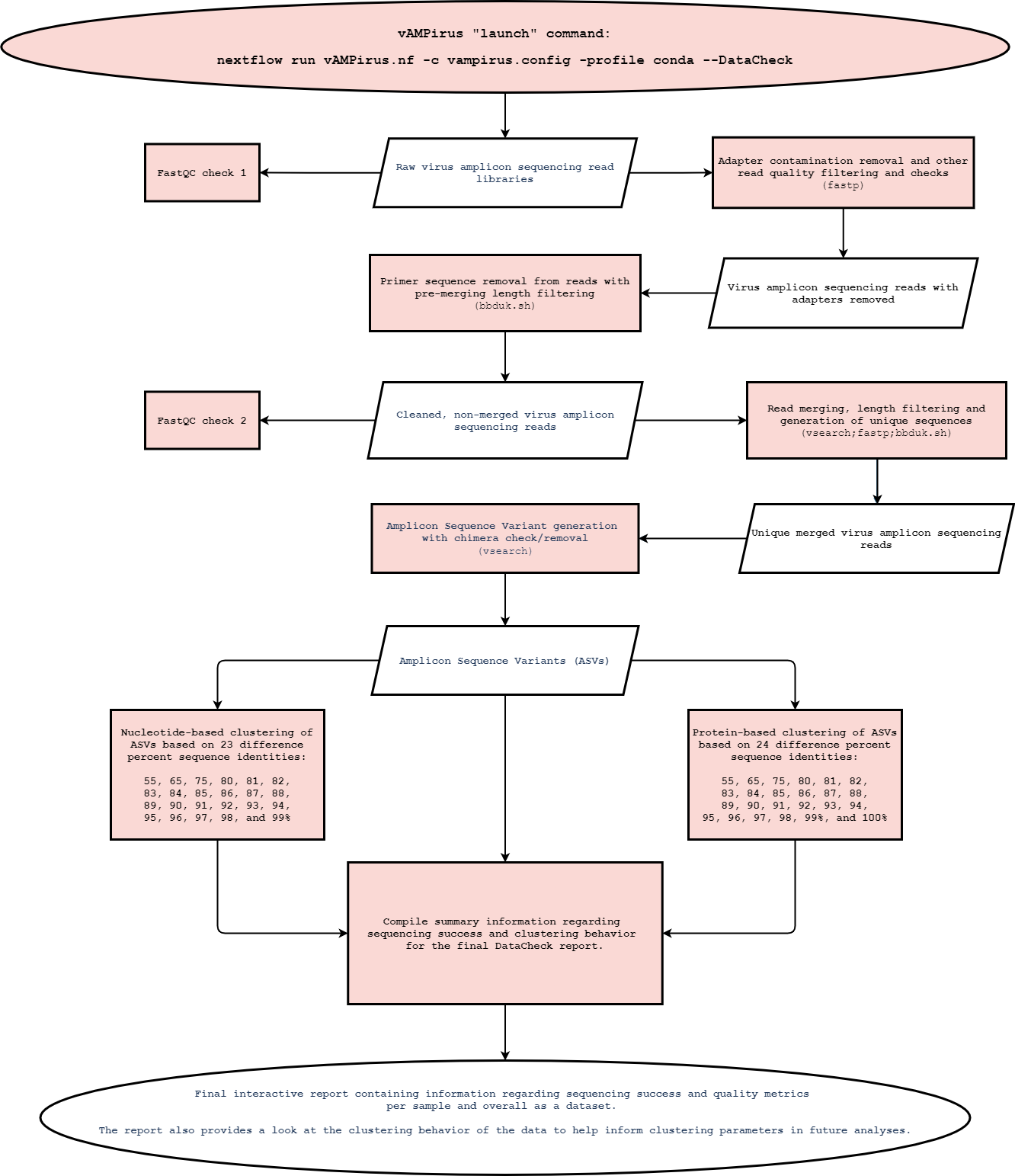
- DataCheck mode ~Click here for flowchart~ -> A preliminary glimpse into dataset sequencing quality and clustering behavior. Provides users with an interactive report with information regarding sequencing success and clustering behavior. This pipeline is meant to help researchers decide appropriate parameters for their analysis by reviewing important characteristics of their dataset.
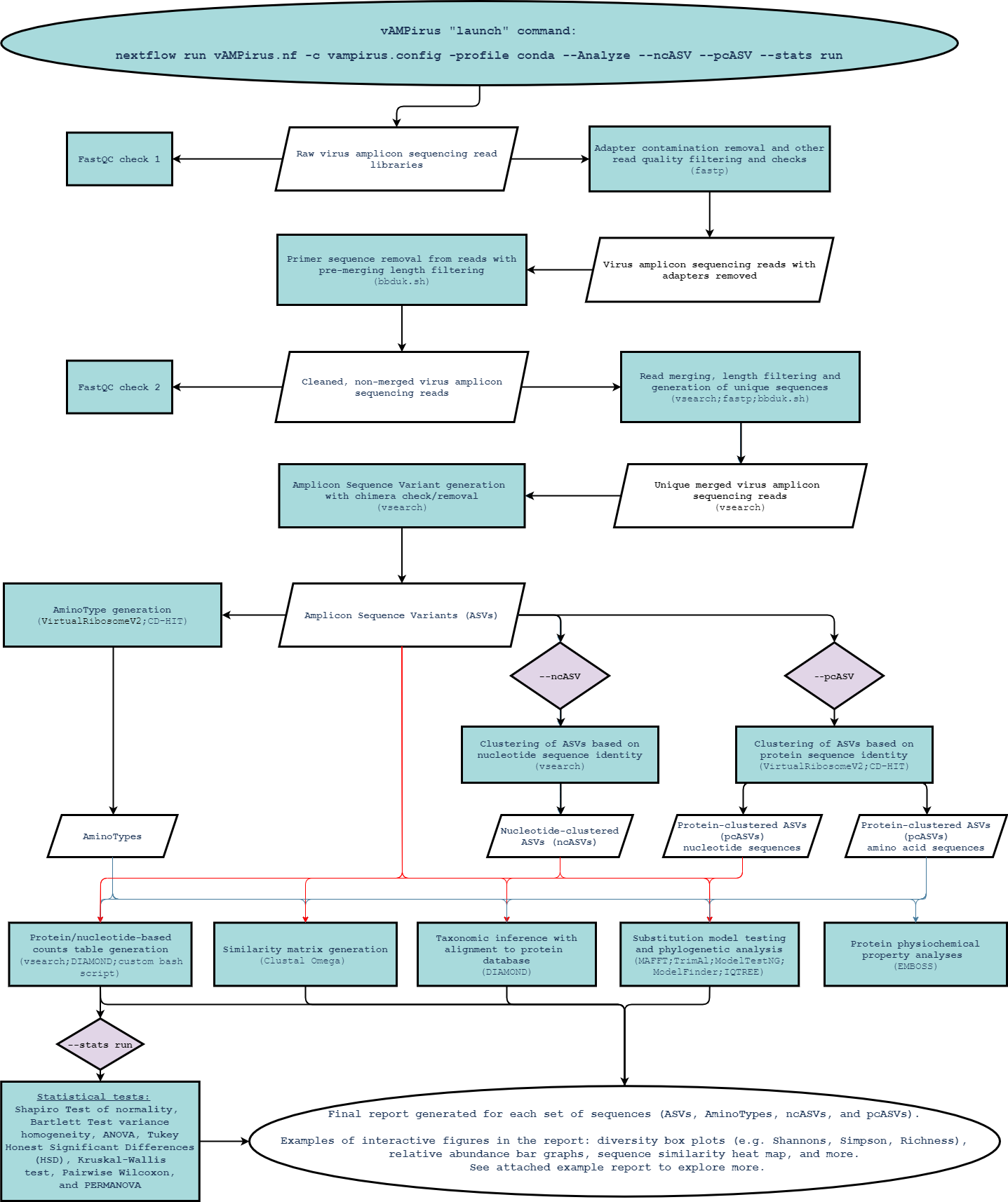
- Analyze mode ~Click here for flowchart~ -> A comprehensive amplicon sequencing analysis pipeline with taxonomy inference, substitution model testing, phylogenetic analyses, protein physiochemical property analyses, and more.
{kind=link}
{kind=link}
Example reports generated by vAMPirus
Reports contain interactive tables and figures that can be downloaded as a .svg file.
1. Download example DataCheck report
2. Download example Analyze report
**These reports were produced with a very small test dataset which may lead to odd looking results (e.g. failed NMDS)
The dependencies
-
DIAMOND v0.9.30 - Buchfink B, Xie C, Huson DH. (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods. 12(1):59-60. doi:10.1038/nmeth.3176
-
FastQC v0.11.9 - Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
fastp v0.20.1 - Chen, S., Zhou, Y., Chen, Y., & Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 34(17), i884-i890.
-
Clustal Omega v1.2.4 - Sievers, F., Wilm, A., Dineen, D., Gibson, T.J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Söding, J. and Thompson, J.D., 2011. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular systems biology, 7(1), p.539.
-
IQ-TREE2 v2.0.3 - Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., & Lanfear, R. (2020). IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution, 37(5), 1530-1534.
-
ModelTest-NG v0.1.6 - Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., & Flouri, T. (2020). ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Molecular biology and evolution, 37(1), 291-294.
-
MAFFT v7.446 - Katoh, K., & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution, 30(4), 772-780.
-
vsearch v2.14.2 - Rognes, T., Flouri, T., Nichols, B., Quince, C., & Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ, 4, e2584.
-
BBMap v38.79 - Bushnell, B. (2014). BBTools software package. URL http://sourceforge. net/projects/bbmap.
-
trimAl v1.4.1 - Capella-Gutiérrez, S., Silla-Martínez, J. M., & Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics, 25(15), 1972-1973.
-
CD-HIT v4.8.1 - Fu, L., Niu, B., Zhu, Z., Wu, S., & Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28(23), 3150-3152.
-
EMBOSS v 6.5.7.0 - Rice, P., Longden, I., & Bleasby, A. (2000). EMBOSS: the European molecular biology open software suite.
-
seqtk v1.3 - Li, H. (2012). seqtk Toolkit for processing sequences in FASTA/Q formats. GitHub, 767, 69.
-
UNOISE algorithm - R.C. Edgar (2016). UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing, https://doi.org/10.1101/081257
Files
Aveglia/vAMPirus-v1.0.1.zip
Files
(16.0 MB)
| Name | Size | Download all |
|---|---|---|
|
md5:118b46faefd1d39b0e836cbae1cb2c2c
|
16.0 MB | Preview Download |
Additional details
Related works
- Is supplement to
- https://github.com/Aveglia/vAMPirus/tree/v1.0.1 (URL)